1) Validation의 실시절차
우수품질의 의약품을 제조하기 위해서는 품질과 제조공정의 설계와 확인이 과학적 근거와 타당성을 가지고 엄밀하게 이루어져야 한다.
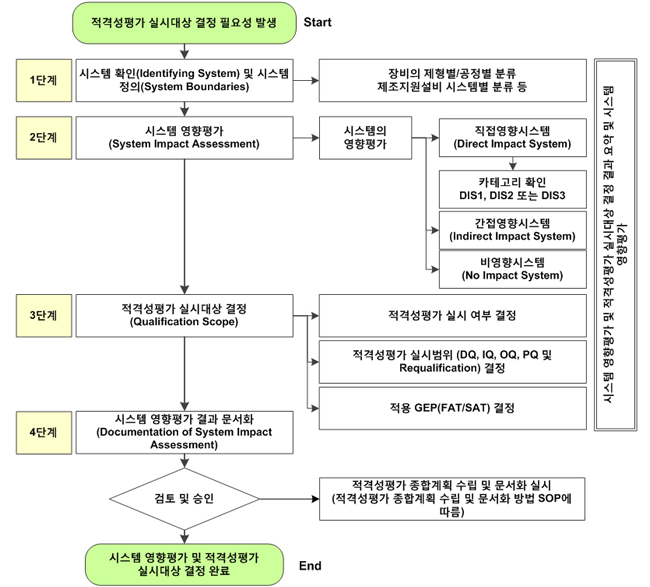
하나의 설비, 공정 또는 방법이 소기의 목적을 달성하고 있는지를 검증·확인하는 validation은 다음과 같은 절차로 실시한다.
① Validation의 실시대상을 선택하고 목적을 분명히 한다.
② 목표로 하는 품질기준을 설정한다.
③ 공정의 특성을 확인하고 가동조건을 결정한다.
④ Validation실시계획서를 작성한다.
⑤ 교정을 수행하여 계측기기를 보정시험하거나 적격성확인을 한다.
적어도 3 제조단위(또는 3회 반복)를 실시하며「최악의 조건」에서도 확인한다..
⑥ 실시 데이터를 수집하여 정리하고 분석한다.
⑦ 결과를 종합적으로 평가해서 문서화한다.
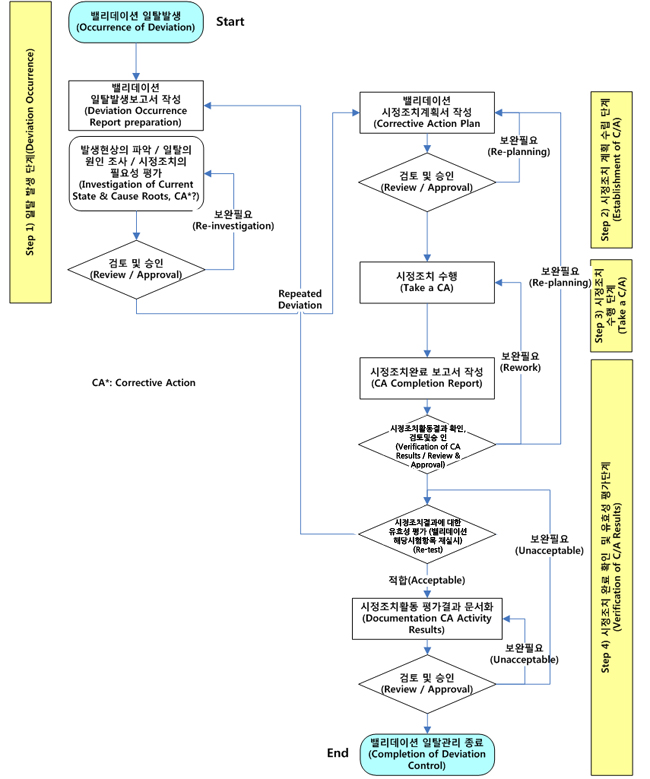
결과가 만족하지 않을 때는 조건을 재설정하여 위의 과정을 반복한다.
이와 같이 실시결과를 종합적으로 평가했을 때 최신의 과학기술 수준에 비추어 만족한 결과가 얻어졌다고 판단되면 그 시스템은 ‘validation되었다’ 라고 할 수 있다.

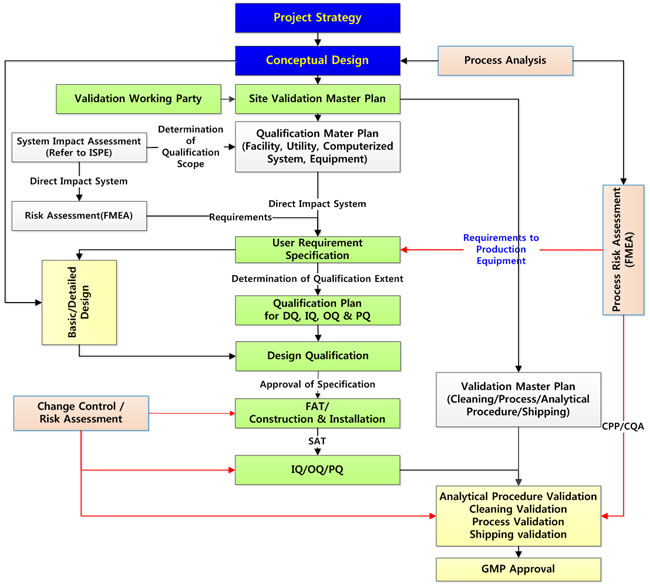
2) 설비의 적격성확인(Qualification) 절차
제조설비, 생산지원설비 또는 시험기기를 구입하여 설치할 때는 설계대로 제작되어 설치되었는지(IQ), 설계대로 가동하는지(OQ), 그리고 소기의 목적을 달성하는지(PQ)를 순서대로 확인하게 된다.